Bacterial Genomics

![]()
![]()
![]()
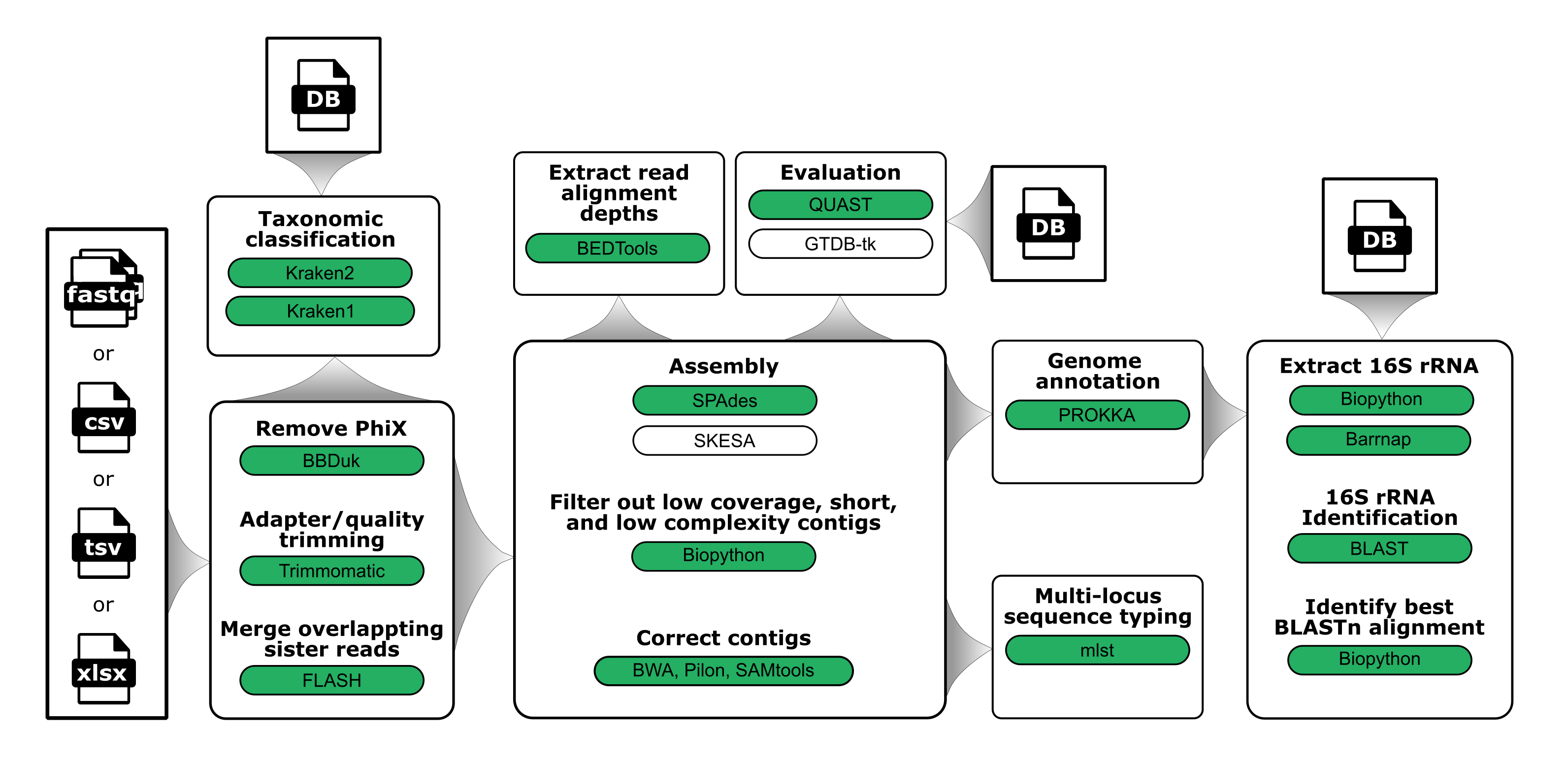
General schematic of the steps in the workflow
Contents
- Quick Start
- Introduction
- Installation
- Usage
- Parameters
- Resource Managers
- Output
- Troubleshooting
- Contributions and Support
- Citations
Quick Start: Test
Run the built-in test set to confirm all parts are working as-expected. It will also download all dependencies to make subsequent runs much faster.
nextflow run \
bacterial-genomics/wf-paired-end-illumina-assembly \
-r main \
-profile singularity,test
Quick Start: Run
Example command on FastQs in “new-fastq-dir” data using SPAdes with singularity:
nextflow run \
bacterial-genomics/wf-paired-end-illumina-assembly \
-r main \
-profile singularity \
--input new-fastq-dir \
--outdir my-results \
--assembler spades
Example command on FastQs in “new-fastq-dir” data using SKESA with singularity:
nextflow run \
bacterial-genomics/wf-paired-end-illumina-assembly \
-r main \
-profile singularity \
--input new-fastq-dir \
--outdir my-results \
--assembler skesa
Introduction
This workflow assembles bacterial isolate genomes from paired-end Illumina FastQ files. Post-assembly contig correction is performed, and a variety of quality assessment processes are recorded throughout the workflow.
This procedure can be used for all bacterial isolates (i.e., axenic, non-mixed cultures) sequenced with whole genome (WGS) or selective whole genome (SWGA) library preparation strategies. It is inappropriate for metagenomics analysis. The data files must be paired read sets (not single ended) and can come from any Illumina sequencing instrument which generates a FastQ file format (e.g., iSeq, HiSeq, MiSeq, NextSeq, NovaSeq).
The read set files can be obtained from an external source, local storage device, or sequencing instrument. Other sequencing manufacturers such as Ion Torrent, PacBio, Roche 454, and Nanopore generate data files that cannot be directly used with this procedure.
[!CAUTION] Due to the shorter DNA fragments of Illumina instruments, reads may be processed as singletons. It is recommended to check the DNA fragment length distribution using TapeStation or BioAnalyzer before sequencing.
Installation
- Nextflow
>=21.10.3 - Docker or Singularity
>=3.8.0 - Conda is currently unsupported
Usage
nextflow run \
bacterial-genomics/wf-paired-end-illumina-assembly \
-r main \
-profile <docker|singularity> \
--input <input directory|samplesheet> \
--outdir <directory for results> \
--assembler <spades|skesa>
Please see the usage documentation for further information on using this workflow.
Parameters
Note the “--” long name arguments (e.g., --help, --input, --outdir) are generally specific to this workflow’s options, whereas “-” long name options (e.g., -help, -latest, -profile) are general nextflow options.
These are the most pertinent options for this workflow:
Required parameters
============================================
Input/Output
============================================
--input Path to input data directory containing FastQ assemblies or samplesheet.
Recognized extensions are: .fastq and .fq with optional gzip compression (.gz)
--outdir The output directory where the results will be saved.
============================================
Container platforms
============================================
-profile singularity Use Singularity images or auto-convert Docker images to run the workflow.
-profile docker Use Docker images to run the workflow.
============================================
Optional assemblers
============================================
--assembler Specify which assembler to execute (spades, skesa). [Default: spades]
============================================
Reference files
============================================
--phix_reference Path to PhiX reference file in FastA format.
Recognized extensions are: (fasta|fas|fa|fna). [Default: PhiX_NC_001422.1.fasta]
--adapters_reference Path to adapter reference file in FastA format. Recognized extensions are: (fasta|fas|fa|fna).
[Default: dapters_Nextera_NEB_TruSeq_NuGEN_ThruPLEX.fas]
PhiX reference NC_001422.1 can be obtained from NCBI.
Optional parameters
============================================
Optional databases
============================================
--kraken1_db Path to a local directory, archive file, or a URL to a compressed tar archive
that contain files `database.{idx,kdb}` and `taxonomy/{names,nodes}.dmp`.
[Default: MiniKraken 8GB]
--kraken2_db Path to a local directory, archive file, or a URL to a compressed tar archive
that contain `{hash,opts,taxo}.k2d` files.
[Default: Kraken2 Standard 8GB]
--blast_db Path to a local directory, archive file, or a URL to a compressed tar archive
that contains BLAST 16S ribosomal RNA files.
[Default: NCBI's 16S ribosomal RNA database]
--gtdb_db Path to a local directory, archive file, or a URL to a compressed tar archive
that contains the GTDB database.
[Default: NaN]
--busco_db Path to a local directory, archive file, or a URL to a compressed tar archive
that contains BUSCO lineages. Can either be a lineage dataset or entire BUSCO database.
[Default: NaN]
[!NOTE] If user does not specify inputs for parameters with a default set to
NaN, these options will not be performed during workflow analysis.
Additional parameters
View help menu of all workflow options:
nextflow run \
bacterial-genomics/wf-paired-end-illumina-assembly \
-r main \
--help \
--show_hidden_params
Resource Managers
The most well-tested and supported is a Univa Grid Engine (UGE) job scheduler with Singularity for dependency handling.
- UGE/SGE
- Additional tips for UGE processing are here.
- No Scheduler
- It has also been confirmed to work on desktop and laptop environments without a job scheduler using Docker with more tips here.
Output
Please see the output documentation for a table of all outputs created by this workflow.
Troubleshooting
Q: It failed; how do I find out what went wrong?
A: View file contents in the <outdir>/pipeline_info directory.
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
Citations
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.