Bacterial Genomics
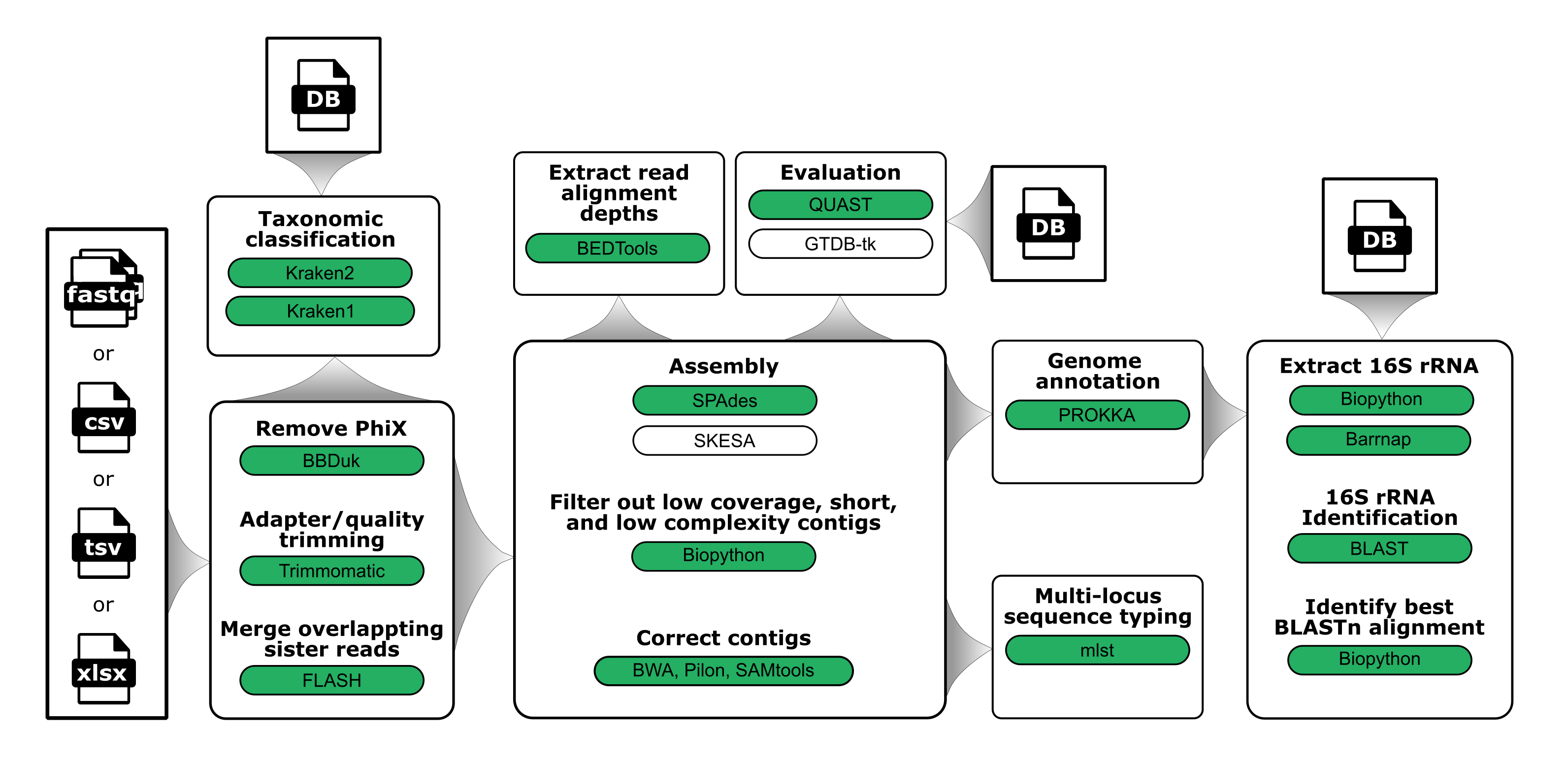
General schematic of the steps in the workflow
Requirements
Nextflow(>=22.04.3)DockerorSingularity(>=3.8.0)
Install workflow locally
git clone https://github.com/bacterial-genomics/wf-paired-end-illumina-assembly.git
Run workflow
Before running workflow on new data, the workflow should be run on the built-in test data to make sure everything is working properly. It will also download all dependencies to make subsequent runs much faster.
cd wf-paired-end-illumina-assembly/
nextflow run main.nf -profile singularity,test --outdir results
Usage
nextflow run main.nf \
-profile singularity \
--input INPUT_DIRECTORY \
--outdir OUTPUT_DIRECTORY \
--assembler <spades|skesa>
When running locally, --max_cpus and --max_memory may need to be specified. Below, max CPUs is set to 4 and max memory is set to 16 (for 16GB).
nextflow run main.nf \
-profile singularity \
--input INPUT_DIRECTORY \
--outdir OUTPUT_DIRECTORY \
--max_cpus 4 \
--max_memory 16
Help menu of all options
nextflow run main.nf --help
Test data was generated by taking top 1 million lines (=250k reads) of SRA data SRR16343585. (Note: This requires SRA Toolkit)
fasterq-dump SRR16343585
head -1000000 SRR16343585_1.fastq > test_R1.fastq
head -1000000 SRR16343585_2.fastq > test_R2.fastq
pigz test_R{1,2}.fastq